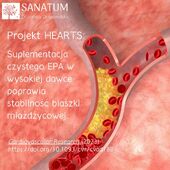
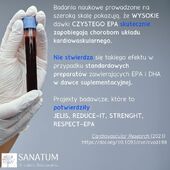
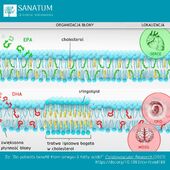
Suplementy diety sklep internetowy Naturopatia-Sanatum.pl – suplementy diety najwyższej jakości dostępne online!
Suplementy diety to preparaty zawierające witaminy, minerały i inne substancje odżywcze, które w korzystny sposób wpływają na procesy fizjologiczne w naszym organizmie. Zadaniem suplementów diety jest uzupełnienie naszej podstawowej diety. Podstawą utrzymania dobrego zdrowia, jak również umożliwienia organizmowi walki ze schorzeniami jest odpowiednia dieta i zdrowy tryb życia. Suplementy diety w żadnym wypadku nie mogą zastępować zdrowego, bogatego w substancje odżywcze i witaminy pożywienia. Jednak istnieje wiele powodów i sytuacji, w których suplementy diety są niezastąpionym uzupełnieniem diety. Należy pamiętać, że suplement diety to nie jest lek, dlatego nie warto wierzyć w reklamy i myśleć, że posiada on takie właściwości. Co ważne sam fakt nazwania preparatu z witaminami lekiem nie gwarantuje tego, że będzie on skuteczny. To, co zapewnia skuteczność suplementu diety w uzupełnianiu potrzebnych składników odżywczych to ich forma, dawka, pochodzenie, a także odpowiednie opakowanie i najwyższej jakości proces produkcyjny. Suplementy diety dostępne w naszym sklepie internetowym zawierają aktywne składniki, charakteryzują się najwyższą jakością i czystością oraz są pozyskiwane z bezpiecznych źródeł. W trakcie ich produkcji zachowane są najwyższe standardy, a produkt końcowy podlega stałej kontroli. Nasi klienci pragnący zadbać o swoje zdrowie mogą czuć się u nas w pełni bezpieczni. Niestety żyjemy w czasach, gdy ilość cennych składników mineralnych oraz witamin w produktach spożywczych maleje. Z tego powodu odpowiednio dobrana suplementacja jest niezwykle ważna.Naturopatia-sanatum sklep z suplementami diety powstał z myślą o osobach dbających o zdrowie, dobrą formę oraz samopoczucie. W naszej ofercie dostępnych jest wiele naturalnych suplementów diety najwyższej jakość. Znajdziesz tu zarówno produkty dla seniorów, dzieci, kobiet w ciąży, jak i suplementy i witaminy niezbędne podczas menopauzy. Nasze suplementy posiadają szerokie zastosowanie – bez najmniejszego problemu znajdziesz u nasz suplement zawierający składniki, które wspomogą Twoją pamięć, zdrowy sen, pracę mózgu, czy dobry nastrój. Nie brakuje u nas również suplementów ze składnikami potrzebnymi pięknej i zdrowej skórze, włosom paznokciom, czy ze składnikami pomagającymi utrzymać równowagę hormonalną. Także osoby borykające się z problemami z tarczycą, stawami, kośćmi, sercem, odpornością czy stanami zapalnymi znajdą suplementy z witaminami, minerałami i innymi substancjami odżywczymi odpowiednimi dla siebie. Jeśli czujesz się zagubiony wśród szerokiej gamy produktów, nasz sklep suplementy diety posiada eksperta, który w mig znajdzie coś idealnego dla siebie. Przekonaj się sam, jak to jest być dobrze zaopiekowanym!
Sklep z suplementami diety i witaminami dla każdego!
Wiele osób zadaje sobie pytanie – dlaczego warto stosować suplementy diety? Śpieszymy z odpowiedzią! Odpowiednio dobrana suplementacja jest niezwykle istotna. Warzywa i owoce, które przepełniają sklepowe półki, niestety nie posiadają już tylu witamin, co to, które były uprawiane kilkadziesiąt lat temu. Związane jest to głównie ze zmianą sposobów uprawy roślin i wprowadzeniem sztucznych nawozów. Dodatkowo brak dostępu do zdrowej żywności oraz ciągły pośpiech sprawia, że niestety bardzo często sięgamy po silnie przetworzoną żywność, która tak naprawdę poza kaloriami nie dostarcza naszemu organizmowi nic pożytecznego. Suplementy diety sklep online, taki jak nasz, oferuje suplementy i minerały, które są pierwszą linią obrony przed niedoborami ważnych dla zdrowia składników. Stawiamy na bogaty wybór wyłącznie najlepszych suplementów diety, które posiadają w swoim składzie wiele wartościowych substancji. Doskonale zdajemy sobie sprawę z tego, że problemy każdego z nas są zróżnicowane. Przecież prowadzimy inny tryb życia, jesteśmy w różnym wieku i posiadamy zupełnie inne problemy zdrowotne. Pragniemy, aby każdy z Was miał w swoim domu suplementy i witaminy sklep najwyższej jakości i mógł odpowiednio wspierać zdrowie swoje oraz swojej rodziny. Naturalne suplementy sklep Naturopatia-Sanatum czeka właśnie na Ciebie!
Suplementy diety online – sposób na wygodne zakupy
Nie da się ukryć, że sklep online suplementy diety to najwygodniejsza forma robienia zakupów. Możesz o każdej porze dnia i nocy zamówić najlepsze, naturalne suplementy prosto pod drzwi swojego domu! Wybierzesz u nas dogodny dla Ciebie sposób dostawy i płatności. Zdrowie to najcenniejsze, co posiadamy, dlatego nie czekaj i już teraz weź je w swoje ręce! Jako sklep internetowy Suplementy Diety zrobimy wszystko, aby zaopatrzyć Cię w najlepsze oraz, co bardzo ważne, pochodzące z pewnego źródła witaminy, minerały i inne ważne składniki. Doskonale zdajemy sobie sprawę z tego, że w obecnych czasach dostępny jest ogromny wybór suplementów diety. Chcąc wzbogacić swoją dietę o właściwą suplementację, powinieneś zainteresować się naszymi produktami. Czym tak właściwie jest właściwa suplementacja? Otóż niczym innym niż dostosowaną do Twoich potrzeb dawką aktywnych substancji odżywczych odpowiednio skomponowanych, które pomogą Ci uzupełnić Twoją dietę! Nieprawidłowa suplementacja, suplementacja niskiej jakości lub nieodpowiednio dopasowana do potrzeb może nie tylko nie być skuteczna, ale także nam zaszkodzić. Warto wspomnieć, że suplementacja nie zastępuje prawidłowego odżywiania, a jedynie je uzupełnia. Gwarantujemy, że nasz sklep z suplementami diety pomoże Ci w walce o Twoje zdrowie!